The Laboratory for Biomolecular Simulation Research made a big impact at the recent 255th American Chemical Society National Meeting in New Orleans, LA (Mar. 18-22, 2018). Special highlights include:
- A special session in the BIOL Division on "RNA Structure and Function" that focused on the predictive understanding of RNA enzymes featured talks by an interdisciplinary research team involving the labs of Phil Bevilacqua (Penn. State), Michael Harris (U. Florida), Joseph Piccirilli (U. Chicago) and anchored by the lab of Darrin York (Rutgers).
- LBSR research faculty Tai-Sung Lee and David Cerutti received prestigious Nvidia GPU Computing Awards for their pioneering efforts to develop exceptional high performance simulation tools on graphical processing units.
- In collaboration with the Rutgers Cyberlearning Innovation and Research Center (CIRC) and the Purdue Institute for Drug Discovery, COMP division poster presentations were enhanced by augmented and virtual reality elements that were highlighted in C&EN News Live Facebook Event.
- Special highlights in Chemistry Education Research were made by graduate student Emily Atieh who made both oral and poster presentations at the meeting.
[ LBSR presented 14 posters/talks this year. Click here for the full ACS agenda for LBSR ]
Following the meeting in New Orleans ACS meeting, York lab members had a follow-up retreat in Florida to engage in team building and discuss research goals and milestones for the upcoming year. [Click Here for more information]
|
|
 Tai-Sung Lee and David Cerutti wins the Tai-Sung Lee and David Cerutti wins theNvidia-Sponsored GPU Computing award --Read More-- |
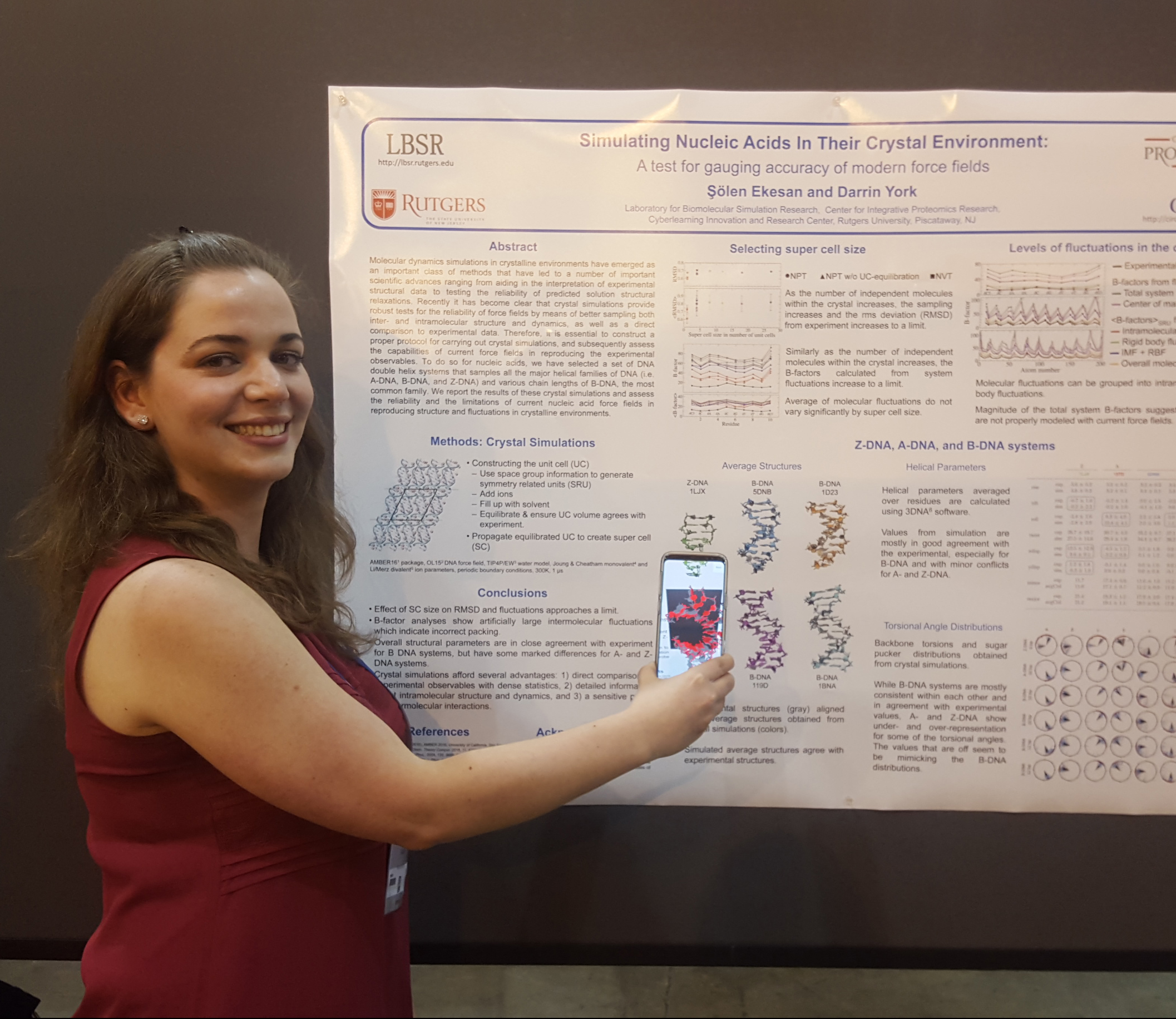 York Research Group (Abir, Colin, Ken, Şölen) uses York Research Group (Abir, Colin, Ken, Şölen) usesaugmented reality during COMP poster session --Read More--
|
 Emily Atieh presents talk and poster Emily Atieh presents talk and posterabout the future of ChemED --Read More-- |
 Special Session:
Special Session:
RNA Structure & Function
![]() Insights into the diverse array of catalytic strategies of RNA enzymes from computational enzymology
Insights into the diverse array of catalytic strategies of RNA enzymes from computational enzymology
Darrin M York
In Biology, RNA enzymes, or ribozymes, play important roles in gene regulation, RNA processing, and peptide synthesis. However, the function of many ribozymes have yet to be unveiled, and new ribozymes continue to be discovered. Recently, there has been a surge of progress in the identification of new classes of self-cleaving nucleolytic ribozymes that have been revealed by comparative genomics analysis, and characterized structurally by X-ray crystallography. However, crystal structures often are not representative of the active states in solution due to a number of factors, and the unified mechanistic interpretation of experimental structural and functional data requires detailed molecular simulations and computational modeling. The present talk will focus on describing a comprehensive computational enzymology approach to study RNA catalysis, with specific applications to unveiling the diverse mechanistic strategies of nucleolytic ribozymes. Molecular simulation models, including ab initio combined quantum mechanical/molecular mechanical (QM/MM) methods as well as fully quantum mechanical force fields (QMFFs) and GPU-accelerated free energy methods are used to study the many facets of RNA catalysis for a series of nucleolytic ribozymes including the hammerhead, VS, pistol, twister, and TS ribozymes. These studies reveal common themes and new twists in the diverse array of catalytic strategies employed by RNA enzymes, and highlight current challenges and important future directions in the field.
Computational RNA enzymology approach that utilizes multiscale quantum modeling methods to unify the interpretation of experimental biochemical and structural data to arrive at a detailed atomistic description of catalytic mechanism.
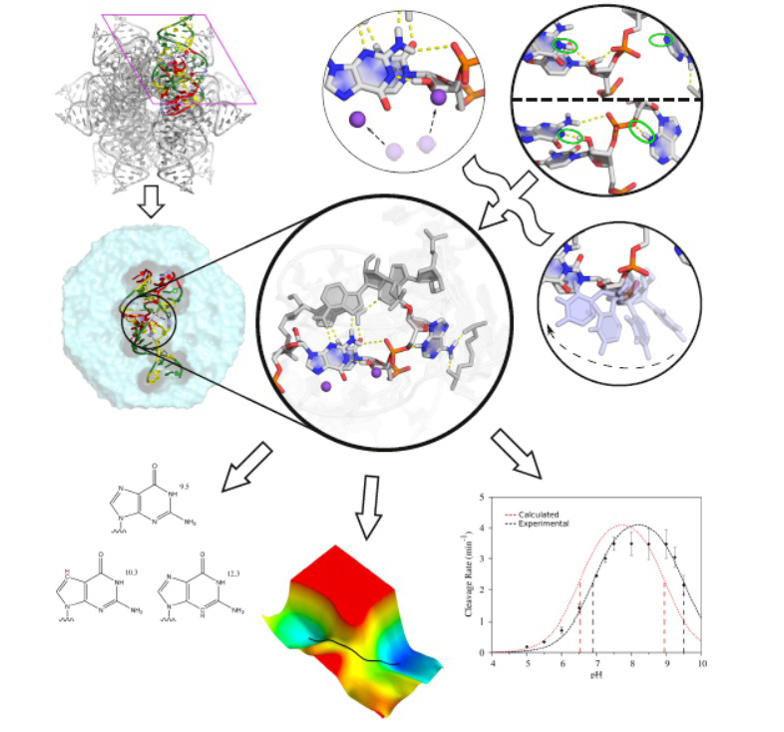
![]() Computational RNA enzymology: Unravelling the catalytic mechanism of the twister ribozyme
Computational RNA enzymology: Unravelling the catalytic mechanism of the twister ribozyme
Colin Gaines & Darrin M. York
There has been a recent flurry of research on the identification and characterization of a number of new small self-cleaving ribozymes, most notably, the twister ribozyme. In addition to a number of crystallographic structures, a wide array of biochemical experiments have been performed and yet a unified model for the active state in solution remains elusive. Here we present a holistic approach to computational RNA enzymology and its application to the twister ribozyme, in order to develop such a unified model that is consistent will all of the available experimental data. First and foremost, a careful analysis of each currently available crystallographic structure was performed in an effort minimize the impact of crystal packing contacts and deactivating mutations on simulations of the active ribozyme in solution. Next a wide array of GPU-accelerated molecular dynamics simulations and free energy calculations were completed and allowed for the development of a mechanistic model consistent with the available experimental data. These simulations explored the structure and dynamics of the twister ribozyme in solution at various points along the reaction pathway, and provided predictions for the pKa of active site nucleobases directly participating in catalysis. The reaction kinetics of a variety of mechanistic models were then explored with ab initio QM/MM simulations and a number of experimentally testable predictions (e.g. kinetic isotope effects, mutations of the catalytic nucleobases, etc.) were developed. In summary, we present the first end-to-end computational study of a small nucleolytic ribozyme and highlight the need for close integration of theory and experiment.
Tai-Sung Lee and David Cerutti wins the Nvidia-Sponsored GPU Computing award
LBSR members Tai-sung Lee and David Cerutti won NVIDIA GPU Award for Best GPU Poster at the 255th ACS conference in New Orleans. This is the first time this award was given to two people. They each received an NVDIA TITAN V GPU.
| GPU-accelerated alchemical free energy simulations |
PMEMD18: High Performance Molecular Dynamics made accessible to scientific programming |
 |
 |
| Tai-Sung Lee <<View Poster>> |
David Cerutti <<View Poster>> |
York Research Group (Abir, Colin, Ken, Şölen) uses augmented reality during COMP poster session
At this meeting, the COMP division will be showcasing a new form of research "posters" that are enhanced with augmented reality (AR) and virtual reality (VR) elements that enable interactive 3D molecular visualization and provide an immersive experience. This work involved a multi-institutional collaboration between the laboratory for Biomolecular Simulation Research (LBSR) and the Cyberlearning Innovation and Research Center (CIRC) at Rutgers and the Purdue institute for Drug Discovery. The AR app was developed by the CIRC team (James Dipierro and James Chun) and showcased on the Chemical & Engineering News FaceBook Live.

| Darrin speaks about the advantages of augmented realtiy | See an interactive augmented reality at a poster session |
|
COMP Poster presentation featuring Augmented Reality |
|
 Şölen Ekesan |
Simulating Nucleic Acids in Their Crystal Enviroment Molecular dynamics simulations in crystalline environments have emerged as an important class of methods that provide robust tests for the reliability of force fields by means of better sampling both inter- and intramolecular structure and dynamics, as well as a direct comparison to experimental data. Therefore, it is essential to construct a proper protocol for carrying out crystal simulations, and subsequently assess the capabilities of current force fields in reproducing the experimental observables. To do so for nucleic acids, we have selected a set of DNA double helix systems that samples all the major helical families of DNA (i.e. A-DNA, B-DNA, and Z-DNA) and report the results of these crystal simulations and the limitations of current nucleic acid force fields in reproducing structure and fluctuations in crystalline environments. |
 Abir Ganguly |
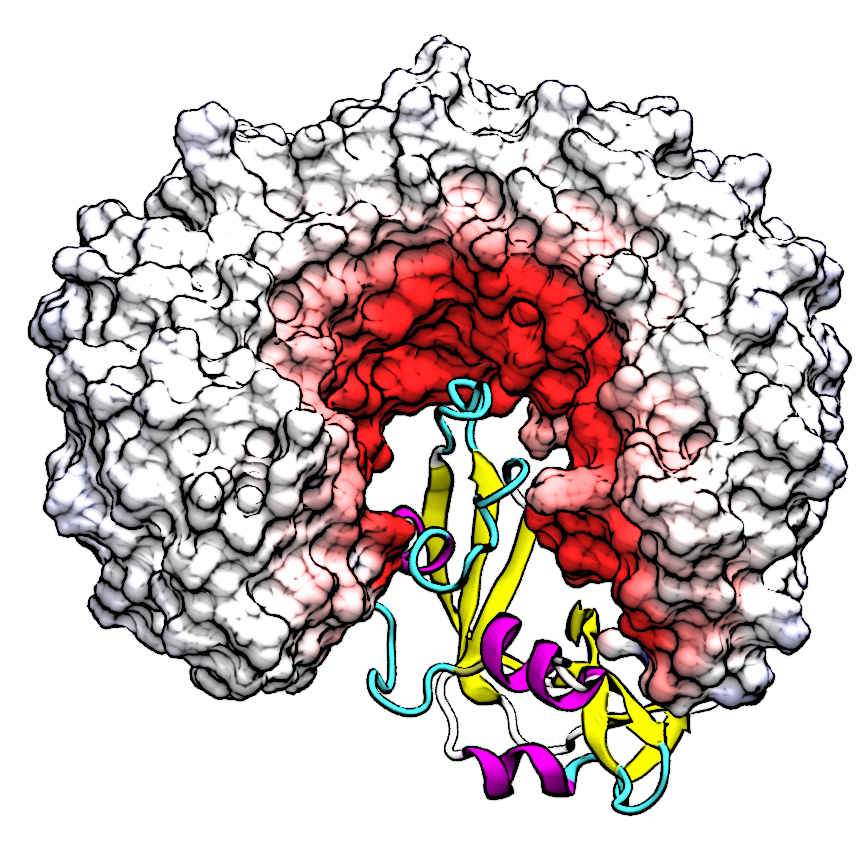
Catalysis in the Varkud Satellite Ribozyme The Varkud Satellite ribozyme (VSr), a member of the nucleolytic ribozyme family that currently also includes the Hammerhead, Hairpin, HDV, glmS, Twister, Twister Sister, Pistol, and Hatchet, catalyzes site specific RNA backbone cleavage reactions that are typical to these small, self-cleaving RNAs. Despite a long history of experimental studies spanning nearly 25 years, several aspects of catalysis in VSr remain under speculation. Recent reports of high resolution crystal structures of VSr have now created an opportunity for computational methods to step in. In this work, we use a variety of state-of-the-art computational techniques to come up with a structural and dynamical picture of catalysis in VSr that is consistent with past and ongoing experiments. |
 Colin Gaines |
With a wealth of new ribozymes being discovered and characterized, it was important to develop of a comprehensive approach to the computational study of catalytic RNA. By bringing to bear a wide variety of tools, including Molecular Dynamics, GPU-accelerated TI calculations, and ab initio QM/MM simulations, we show how this method has been successful in developing atomically detailed models for both the active state of the twister ribozyme in solution as well as plausible mechanistic pathways. Further, we have made a number of experimentally testable predictions based on these models and highlight the need for close integration of theory and experiment. |
 Ken Kostenbader |
Two recently discovered ribozymes, called TS and pistol, have been crystallized and biochemically characterized, but questions still remain about the role of magnesium ions in their respective catalytic mechanisms. GPU-accelerated molecular dynamics simulations were employed to help gain insight into central questions concerning the coordination chemistry of these hydrated metal ions in their respective active sites. Insights were gained about the effect of metal binding modes on the four catalytic strategies of the pistol ribozyme. |
|
Additional Presentation by LBSR |
|
|
Gerhard Koenig |
Convergence of (multi-scale) free energy simulations Multi-scale free energy simulations (MFES) based on molecular mechanics (MM) and quantum mechanics (QM) offer a clear pathway to improve the accuracy of free energy results. While simulations at the MM level allow the sampling of all relevant degrees of freedom, the post-processing with QM ensures the proper treatment of molecular interactions. However, just like conventional free energy techniques, MFES approaches only converge if there is sufficient phase space overlap between the MM and the QM potential energy surfaces. We employ harmonic benchmark systems to develop a simple metric to evaluate a priori whether a free energy simulation is likely to converge. In addition, we determine the most problematic MM interaction types with regard to phase space overlap with the QM energy surface. We also compare different ad hoc strategies to mitigate the lack of phase space overlap. |
|
George Giambasu |
Using topology of molecular solvation for biomolecular structural refinement Binding of ions and water to specific sites is intimately linked with how supramolecular assemblies form and how biomolecules fold and behave in a test-tube, crystal or cell. The new models based on 3D-RISM and new topological analysis methods that I developed are powerful computational tools for prediction of site-specific binding to a wide array of biological systems and molecules relevant to supramolecular chemistry. Herein, these new tools are applied to crystallographic refinement of a variety of nucleic acids molecules and ion channels using the the AMBER molecular simulation suite coupled with the PHENIX general purpose crystallographic structure refinement program. |
|
Taisung Lee |
Recently the rapid advance of GPU-accelerated computing has made significant impact on scientific, analytics, engineering, consumer, and enterprise applications. In the area of molecular dynamics (MD) based alchemical free energy simulations, currently the most accurate approaches for ligand binding problems, GPU-accelerated implementation has caught broad attention only very recently. Here we report the advancement of the GPU-accelerated pmemdGTI package for alchemical free energy methods. This package has been integrated into the official AMBER 18 release, and is fully supported with the full spectrum of features in AMBER. Additionally, the package enables full functionality to several new features that are in the AMBER pipeline, but that will not part of the next official release, including new enhanced sampling methods, metal ion potentials and integration with new Parameter-Interpolated Thermodynamic Integration (PI-TI) methods. |




Emily Atieh presents talk and poster about the future of ChemED
Sleuthing Through Chemistry: Finding the Cheapest Route to a Multi-step Problem
Emily L. Atieh, Darrin M. York
It is no secret that students often approach chemistry problems in a rote-memorization fashion. Rather than asking what the problem means, it is more often that our instructors hear “What equation do I use for this problem?” The result of this is that students are more hesitant to solve problems in which the wording or the variables given have been changed. Our goal was to develop problem sets in which students must pick their own variables and develop their own solution maps. These “sleuth problems” provide students with a goal, usually a typical “ask” from a traditional chemistry problem, like finding the threshold frequency of a metal, along with a long list of variables. The trick, however, is that not all variables are created equally. Each variable has a cost associated with it, and a more obvious solution will cost the students more. Beginning with only a limited budget, students must be more creative to search out another, less obvious pathway. We have piloted our sleuth problems using an online format within our active learning recitations. Students work in groups of three to come up with the most cost-efficient solution, while being assisted by our teaching interns. In our preliminary observations, we found that our students had an easier time sticking to the task, asked each other more questions, and took time to reflect on not only their methods, but also their results.

The Undergraduate Certificate in Chemistry Education Program: Updates and Findings
Emily L. Atieh, Darrin M. York
Peer instruction has been a booming topic within STEM education over the last few decades. Gateway courses, such as General Chemistry, have particularly gained our attention due to high enrollments and notorious rates of attrition. At Rutgers University, the Chemistry Department hosts the Teaching Internship program. This program provides course credit for our students to become peer instructors within General Chemistry. Through pedagogical training, teaching, and reflection, teaching interns are trained to put theory into practice and practice into learning. They work within our courses to fill some traditional needs, like holding office hours or review sessions. They also play a large role in our shift towards active learning, hosting workshops and assisting in our newly designed recitations. Some teaching interns opt to take on additional coursework and teaching opportunities in order to earn either a Certificate in Chemistry Education or a minor in Chemistry Education. Teaching interns have provided the department with an excellent opportunity to learn more about how teaching and training impacts the peer instructors themselves. In an attempt to paint a full picture of the teaching intern, our goal is to monitor their shifts in attitudes towards learning, their beliefs about teaching and ability to implement best practices, and their metacognitive habits. Results and insight into this role may provide a model for training other peer instructors, as well as graduate students and new faculty.


Darrin M York
BIOL 24: Computational RNA enzymology: Unravelling the catalytic mechanism of the twister ribozyme
Colin Gaines & Darrin M. York
CHED 127: Creation of an undergraduate certificate in chemistry education program: Updates and findings
Emily Lauren Atieh & Darrin M York
CHED 2015: Sleuthing through chemistry: Finding the cheapest route to a multi-step problem
Emily Lauren Atieh & Darrin M York
COMP 205: Catalysis in the Varkud satellite ribozyme
Abir Ganguly & Darrin M York
COMP 215: Computational RNA enzymology: New tools to unravelling the catalytic mechanism of nucleolytic ribozymes
Colin S Gaines & Darrin M York
COMP 228: Investigating the impact of metal binding modes on ribozyme catalysis through the use of GPU-accelerated simulations
Ken Kostenbader & Darrin M York
COMP 244: Simulating nucleic acids in their crystal environment: A test for gauging accuracy of modern force fields
Solen Ekesan & Darrin M York
COMP 322: GPU-accelerated alchemical free energy simulations: A new tool for accurate, affordable prediction of ligand binding potency for drug discovery, optimization, and resistance problems
Tai-Sung Lee & Darrin M York
COMP 378: Quantifying drug resistance at the atomic level through GPU-accelerated affinity simulations
Tai-Sung Lee & Darrin M York
COMP 35: Convergence of (multi-scale) free energy simulations
Gerhard Koenig & Darrin M York
COMP 221: Using topology of molecular solvation for biomolecular structural refinement
George M Giambasu
COMP 397: Predicting site bound ions and water to nucleic acids using molecular solvation theory
George M Giambasu